
| Le menu « graphes » (mode extraction) | Sommaire | Le menu « EXAFS » (mode extraction) |
Ce menu regroupe les opérations qui permettent d'obtenir le spectre d'absorption total de l'atome absorbeur ou de l'échantillon, et les options qui travaillent directement sur ces spectres (traitement des seuils).
Cette option permet de récupérer dans LASE les fichiers expérimentaux créés au LURE. Il est possible, grâce à cette option, de récupérer directement les valeurs du coefficient d'absorption, mais aussi des intensités avant et après échantillon, aussi bien en fluorescence (avec un ou plusieurs détecteurs) qu'en absorption (avec ou sans chambre d'étalonnage). La nature des informations à extraire se paramètre à l'aide de la boîte de dialogue qui apparaît.
La partie haute permet d'indiquer la ligne expérimentale sur laquelle ont été enregistrés les spectres, ainsi que l'organisation générale du fichier et la façon dont il convient de le traiter. En-dessous, on peut préciser comment sont agencées les données dans le fichier. Enfin, tout en bas, on trouve les traitements que l'on peut appliquer aux graphes ainsi chargés.
Actuellement, sept formats expérimentaux sont reconnus. On peut choisir ces formats grâce au menu déroulant qui apparaît en cliquant sur le bouton dans lequel est indiqué le format sélectionné. Ces formats sont résumés dans la table ci-dessous.
| Format | Lignes | Commentaire | |
|---|---|---|---|
| LURE/Aquvisu — exafs 13 | DCI : D42, D44 | Transmission, fluorescence ou détection d'électrons, multi-élément reconnu | |
| LURE/DCI D21 — exafs 2 | DCI : D21 | Transmission, fluorescence ou détection d'électrons, multi-élément reconnu, spectres successifs reconnus | |
| Hasylab/Doris | Hasylab : Doris | Fluorescence | |
| NSLS micro-XANES | NSLS : ? | Fluorescence. Fichier organisé en lignes | |
| ESRF / BM01B (SNBL) | ESRF : BM01B | Transmission, fluorescence, spectres successifs reconnus, coupures de faisceau signalées | |
| ESRF / ID21 | ESRF : ID21 | Transmission, fluorescence, spectres successifs reconnus, coupures de faisceau signalées. Les spectres sont détectés par la commande z3escan | |
| ESRF / ID22 | ESRF : ID22 | Transmission, fluorescence, spectres successifs reconnus, coupures de faisceau signalées. Les spectres sont détectés par la commande ascan avec un mouvement energy ou menergy |
Dans tous les cas, LASE essaye de détecter automatiquement le nombre de colonnes et, éventuellement, de spectres contenus dans le fichier. Il est cependant possible de préciser ces valeurs, en cas d'échec, en désactivant les boutons Automatique (pour les colonnes) et Auto. (pour le nombre de spectres) et en indiquant la valeur dans le champ éditable qui s'active alors. Le nombre de points de chaque spectre est déterminé automatiquement ; les spectres comptant moins de trois points sont ignorés. En cas d'anomalie dans le fichier (le plus souvent, cela signale une coupure de faisceau ; parfois, une saturation d'un détecteur), LASE affiche un message. Il est alors possible d'ignorer l'erreur, le spectre étant alors chargé normalement, de passer au spectre suivant, le spectre étant alors tronqué, ou d'annuler le chargement du fichier.
Si vous utilisez un autre format de fichier expérimental, il est nécessaire de le convertir dans l'un des deux formats ci-dessus pour que LASE puisse le traiter automatiquement ; s'il est en ASCII, il est aussi possible de charger séparément les différentes intensités avec l'option Importer ASCII puis de calculer le spectre d'absorption à l'aide de l'option Calculer le signal. Vous pouvez aussi essayer de modifier les paramètres associés à un format de fichier déjà connu (nombre de colonnes, caractère introduisant les lignes à ignorer (Commentaire), nombre de lignes à ignorer au début (En-tête).
Si la case Nouvelle série est active, une nouvelle série est créée et les graphes construits à partir des fichiers seront placés dans cette série, sinon ils seront placés dans la série courante (si elle est protégée, une nouvelle série est créée). Si la case Une série par fichier est active, une série sera créée pour chaque fichier sélectionné, sinon les graphes de tous les fichiers seront dans la même série.
Si la case Garder les spectres est active, le logiciel calcule le coefficient d'absorption expérimental à partir des intensités lues dans le fichier.
Cette portion de la boîte de dialogue permet de définir quel traitement appliquer aux valeurs en abscisses, en particulier ce qu'il faut faire en cas de spectres successifs dans le même fichier.
La moitié haute permet de définir la nature des abscisses : la colonne qui les contient, mais aussi leur nature : énergie (en eV ou en keV), angle ou autre grandeur. Si l'énergie est en keV, elle est automatiquement convertie en eV, autrement aucune transformation n'est appliquée.
La moitié inférieure permet d'indiquer la présence de plusieurs spectres les uns à la suite des autres dans le fichier, en activant la case Spec. successifs. Si elle est inactive, LASE considère qu'un seul spectre est contenu dans le fichier et crée donc un seul graphe par détecteur. Sinon, un graphe est créé pour chaque spectre.
On peut indiquer le nombre de spectres, ou laisser LASE le déterminer. Dans ce cas, il considère que l'on change de spectre quand l'abscisse décroît (retour en arrière dans les énergies) ou s'il rencontre le code correspondant, si le format de fichier le prévoit.
Une fois les graphes de chaque passage créés, il est possible de les moyenner directement, ou de les fusionner, en activant les options correspondantes. Dans ce cas, les graphes originaux sont effacés, seul le graphe final est conservé.
Il est possible de n'utiliser que certains spectres du fichier, en précisant leur numéro avec la syntaxe décrite ici.
Cette portion de la boîte de dialogue permet de définir la configuration des détecteurs : quels sont ceux qui mesurent la fluorescence, la transmission ; lesquels doit-on prendre en compte ;...
Tout en haut, il faut indiquer le numéro de la colonne qui contient les intensités mesurées avant l'échantillon (Faisceau incident : I0 colonne ...). Si le bouton Garder est choisi, un graphe contenant ces intensités est créé, sinon elles servent juste au calcul du coefficient d'absorption expérimental.
Le principe est le même pour les trois types de détecteur : si la case à gauche est cochée, il existe de tels détecteurs dans l'expérience et le signal qu'ils mesurent sera pris en compte. Les colonnes correspondantes sont à préciser, avec la syntaxe des plages de valeur, dans le champ en-dessous. Si le bouton Garder est choisi, un graphe contenant ces intensités est créé, sinon elles servent juste au calcul du coefficient d'absorption expérimental.
En transmission, l'intensité du faisceau est mesurée avant l'échantillon (valeur I0) et après traversée de l'échantillon (valeur I1). On montre que, si l'échantillon est homogène et d'épaisseur e constante, la loi de Beer-Lambert s'applique : I1 = I0 e-µe.
En fluorescence, on mesure l'intensité du faisceau avant l'échantillon (valeur I0) et l'intensité du faisceau de rayons X réémis par fluorescence par l'atome absorbeur (valeur If). Dans le cas d'un échantillon dilué d'épaisseur infinie, on montre que l'on a la relation If = µkI0, où k est une constante positive. Dans les deux cas, la valeur de la constante multiplicative ne joue aucun rôle dans l'utilisation ultérieure de µ : elle est donc arbitrairement fixée à 1.
Si plusieurs détecteurs de même nature sont présents dans le fichier (le plus souvent, mesures en fluorescence à l'aide d'un détecteur multiéléments, mais aussi mesure simultanée d'un échantillon et d'une référence en transmission), il est possible de demander la moyenne automatique des détecteurs conservés, en activant le bouton Moyenner en bas de la région. Bien entendu, les moyennes ne sont faites qu'entre détecteurs de même nature... En revanche, si le bouton Séparer est actif, pour chaque détecteur est conservé un spectre distinct.
En transmission, on peut imaginer deux façons de faire cette moyenne : soit sur les intensités transmises, soit après avoir calculé le coefficient d'absorption expérimental. Ces deux méthodes sont disponibles, en activant les boutons log(S) et S(log), respectivement.
Les boutons en bas de la boîte de dialogue permettent d'activer un traitement du spectre à l'issue du chargement. Ce traitement est fait après les moyennes entre détecteurs pour un même enregistrement, mais avant les moyennes entre enregistrements successifs pour un même fichier.
Si l'option trier est active, les graphes obtenus après extraction sont triés par énergie croissante. Ce tri étant théoriquement déjà effectué lors de l'enregistrement, le nombre d'inversions est supposé faible donc un algorithme simple de tri en place par permutations a été adopté. Si le graphe s'avère très désordonné, il peut être plus avantageux d'utiliser les options de tri par blocs, plus performantes. La majorité des opérations de traitement supposant que les valeurs sont triées par énergie croissante, il est conseillé de laisser cette option activée, même si en pratique les inversions d'énergie sont rares.
Si l'option Moy. doublons est active, les graphes obtenus après extraction sont vérifiés de façon à éliminer les points doubles (déplacement du monochromateur trop faible) ; les différentes valeurs enregistrées pour une même énergie sont remplacées par une valeur unique, leur moyenne. Dans le cas où des doublons ont été rencontrés, leur nombre est indiqué pour information.
La présence de points doubles rend impossible certaines étapes du traitement, il est donc conseillé de garder cette option active lors du travail sur un seul spectre. Mais, attention, la présence de points multiples perturbe le traitement statistique des spectres. En effet, lors de deux enregistrements successifs, les points doubles n'apparaissent pas nécessairement à la même énergie, ni même en même nombre. Moyenner les points doubles conduit donc à des décalages en énergie entre les spectres (le i-ème point du spectre 1 n'est pas à la même énergie que celui du spectre 2), voire à des spectres ne contenant pas le même nombre de points. Cela rend délicates les options de moyenne des spectres, qui supposent implicitement l'égalité (aux variations expérimentales près) des abscisses pour chaque point du spectre. Deux alternatives sont possibles : d'une part, après élimination des doublons, demander l'interpolation (ou le recalage) de chaque spectre ; d'autre part, sans élimination des doublons, réaliser une pseudo-fusion qui, au contraire, tire parti des points doubles pour estimer l'erreur (voir le calcul des barres d'erreur).
Si l'option Normaliser est active, les spectres chargés sont automatiquement normalisés, de telle sorte que le maximum vaille 1 et le minimum 0 (cela revient donc à utiliser la normalisation classique). Cette normalisation est la plus simple possible, mais il est préférable de normaliser de telle sorte que la hauteur de seuil soit unitaire : utiliser l'option de normalisation décrite ci-après). Attention ! La normalisation n'affecte que les spectres calculés, pas les valeurs de I0, I1 ou If si elles sont conservées.
Permet de démarrer le traitement des fichiers expérimentaux. Il suffit de choisir un ou plusieurs fichiers dans le sélecteur qui apparaît pour démarrer la conversion. Attention, si vous choisissez plusieurs fichiers, tous sont supposés du même type et sont traités en fonction des paramètres indiqués ci-dessus. Tous les graphes créés sont placés dans une nouvelle série.

Cette option permet de calculer le spectre d'absorption à partir des intensités mesurées expérimentalement, aussi bien en fluorescence qu'en transmission. La boîte de dialogue qui apparaît permet de choisir quel graphe contient les valeurs de I0 (défileur du haut) et quel graphe contient les valeurs de I1 ou If (défileur du bas). Dans ces défileurs, seuls les graphes sans type ou étant explicitement du type convenable apparaissent ; si aucun graphe de l'un ou l'autre type n'existe, un message d'alerte en informe l'utilisateur et l'appel est annulé. Il n'est possible de sélectionner qu'un seul graphe de type I0 et un seul de type I1.
Si les graphes indiqués pour I0 et If possèdent des barres d'erreur, LASE estime les barres d'erreur sur le spectre calculé à l'aide de ces barres d'erreur. Si l'un des deux graphes (ou les deux) n'a pas de barres d'erreur, et si l'option Estimer erreur est active, les barres d'erreur correspondantes sont estimées en utilisant le modèle de la loi de Poisson.
Dans les deux cas, les erreurs sur le signal calculées sont estimées à l'aide du théorème de propagation des erreurs. Cette estimation suppose que I0 et If puissent être modélisées par des variables aléatoires gaussiennes indépendantes, de moyenne et d'écart-types respectifs (µ1, s1) et (µ2, s2) ; dans ce cas, le théorème de propagation des erreurs indique que le quotient peut être estimé par une variable aléatoire gaussienne de moyenne µ et de variance s2 tels que :

Cette estimation est correcte tant que |µ1| est suffisamment grand et s1 suffisamment petit pour que p(N(|µ1|, s1) < 0) soit négligeable, ce qui est toujours le cas dans les conditions usuelles.
Dans le cas de la transmission, il faut en plus tenir compte de l'effet du logarithme et la formule devient
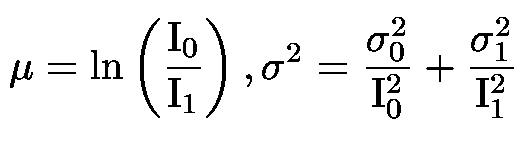
Cette option n'est pas encore disponible.
Cette option n'est pas encore disponible.
Cette option n'est pas encore disponible.
Cette option permet de corriger les énergies d'un spectre, à partir des résultats d'étalonnage avec un composé de référence par exemple. Elle n'a de sens que sur un spectre dont les X correspondent à des énergies exprimées en électronvolts. Cette correction se fait à partir d'un valeur de l'énergie supposée connue théoriquement, dont on indique la valeur attendue Eth (Énergie attendue) et la valeur observée Eobs (Énergie mesurée).
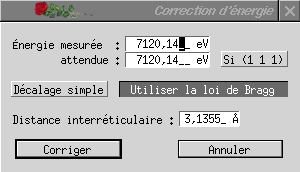
Le décalage simple réalise une correction linéaire des énergies : E' = E + (Eth - Eobs), où E' est l'énergie après correction. Cette correction, très simple, ne tient cependant pas compte du fait que la valeur lue expérimentalement est, en réalité, la valeur de l'angle d'incidence du faisceau sur le cristal du monochromateur.
Utiliser la loi de Bragg permet de tenir compte de ceci : à l'aide de la loi de Bragg, 2dsin(t) = kl où d est la distance entre les plans réticulaires responsables de la réflexion dans le cristal du monochromateur, le programme calcule l'angle d'incidence observé, tobs, et l'angle d'incidence théorique, tth. Les énergies sont ensuite corrigées à l'aide de la formule :

Réaliser cette correction demande de connaître la distance interréticulaire : elle doit être indiquée dans le champ adéquat. Le menu déroulant, à droite de l'énergie attendue, permet d'indiquer un cristal parmi ceux couramment utilisés pour les monochromateurs la distance interréticulaire correspondante est alors proposée. Cette correction n'est pas linéaire, elle conduit à une modification des phases du spectre E.X.A.F.S.
Cette option n'est pas encore disponible.
Cette option permet d'effectuer en une seule fois les différentes étapes usuelles du traitement d'un spectre X.A.N.E.S. expérimental : élimination de l'absorption due à d'autres effets que le seuil étudié, calcul de la dérivée première et de la dérivée seconde pour chercher l'énergie de seuil. Ces opérations sont décrites dans la suite ; elles ne sont effectuées que si le module d'élimination de µm est quitté en confirmant le calcul (voir ci-après).
Lorsque l'enregistrement est fait en transmission, le coefficient d'absorption mesuré est la résultante de l'absorption des rayons X par tous les atomes du composé étudié. Cependant, seul le coefficient d'absorption de l'atome étudié pour la transition étudiée (µ0) est intéressant. Avant tout traitement, il est donc nécessaire de séparer ces deux contributions. Les effets étant additifs, et l'absorption de la transition étudiée étant nulle par définition avant le seuil, cette séparation se fait en soustrayant au spectre d'absorption expérimental µ(E) une fonction qui modélise la résultante de toutes les autres absorptions (fond continu ou absorption de la matrice, µm(E)) : µ0(E) = µ(E) - µm(E). Cette opération est réalisée par cette option.
Cette correction est parfois nécessaire en fluorescence ; dans ce cas, µm représente souvent la contribution du rayonnement diffusé par l'échantillon qui arrive sur le détecteur en plus du rayonnement de fluorescence. En fluorescence, cependant, la correction n'est utile que si le spectre après le seuil présente une pente importante, or cette pente est souvent bien différente de ce que l'on observe avant le seuil : il est donc très rare que cette étape soit utile.
À l'appel de cette fonction, LASE ouvre une fenêtre qui permet de paramétrer le calcul de la fonction µm. Le défileur en haut à gauche permet de choisir le type de fonction à utiliser ; les options à droite contrôlent la façon dont cette fonction est estimée. Le spectre expérimental est en noir, la fonction modélisant µm est en bleu ; le résultat obtenu pour µ0 est tracé en gris.

L'énergie E1 est un paramètre commun à toutes les façons de procéder : il permet de définir la région du spectre qui sert à chercher la meilleure fonction demandée (au sens des moindres carrés). Tous les points situés à une énergie E < E1 sont utilisés pour l'estimation ; la fonction obtenue est ensuite extrapolée à tous les points d'énergie E > E1. La position de E1 est représentée par le trait vertical rouge ; elle peut être modifiée soit en cliquant sur la nouvelle position, soit en entrant une valeur après avoir cliqué sur l'ancienne valeur.
LASE propose quatre types de fonctions pour modéliser µm, avec deux à six paramètres libres. Les valeurs des paramètres sont, dans tous les cas, recherchés de façon à minimiser la distance entre la courbe expérimentale et la fonction modèle (méthode des moindres carrés). Dans tous les cas, les équations des moindres carrés sont linéaires (les fonctions modèles sont linéaires par rapport aux paramètres) : elles sont résolues directement par la méthode du pivot de Gauss.
Pour chacune de ces courbes, le résidu entre le modèle et l'expérience (divisé par le nombre de points utilisés pour le calcul) est affiché en haut, à droite de la fenêtre (Résidu : ...). Dans l'hypothèse où le modèle utilisé pour µm représente correctement la réalité, ce résidu caractérise les fluctuations aléatoires de la mesure et estime donc le carré des erreurs statistiques expérimentales &emdash; ce qui suppose toutefois un grand nombre d'hypothèses : il est préférable, si cela est possible, d'utiliser les techniques d'estimation de l'erreur spécifiques de LASE (moyenne de plusieurs spectres et propagation de l'erreur).
Il arrive que les premiers points d'un spectre soient aberrants. Les prendre en compte gêne donc la détermination de µm. Pour palier ce problème, il est possible, en activant cette option, d'indiquer un second point de contrôle E'1, de telle sorte que seuls les points d'énergie comprise entre E'1 et E1 soient utilisés dans la recherche de µm. La position de E'1 est indiquée par un trait vertical vert ; elle peut être modifiée en cliquant avec le deuxième bouton de la souris à la nouvelle position, ou en entrant la nouvelle valeur après avoir cliqué sur l'ancienne. Si la nouvelle valeur est telle que E'1 > E1, le programme échange les deux valeurs de telle sorte que l'on ait toujours E'1 < E1.
Quand la fonction µm vous satisfait, cliquer sur le bouton de fermeture de la fenêtre : il apparaît un message d'alerte qui vous demande quoi faire (ci-dessous, à gauche).

|

|
Quatre choix sont proposés : Annuler annule la fermeture de la fenêtre, Fermer ferme la fenêtre sans effectuer les calculs. Soustraire réalise la soustraction demandée sur le spectre choisi. Série, en plus de cette soustraction, propose de l'effectuer dans les mêmes conditions sur tous les spectres bruts (donc qui pourraient avoir à être traité ensuite) de la même série.
Dans le cas d'un clic sur Série, la façon dont sont transférées les conditions de reproduction est contrôlée par la seconde boîte d'alerte qui apparaît (ci-dessus, à droite). Avec Identique, c'est exacement la même courbe qui est soustraite. Avec Similaire, les paramètres de cette courbe sont recalculés isolément pour chacune des courbes. Avec Ajuster, les paramètres de cette courbe sont optimisés pour assurer une identité maximale entre les résultats obtenus pour chacun des spectres (distance entre ces résultats minimale au sens des moindres carrés). C'est souvent cette dernière option qui donne les meilleurs résultats.
Si le spectre original possède des barres d'erreur, elles sont conservées pour le spectre corrigé. Cela suppose que les valeurs soustraites (donc les valeurs de µm) sont exactes, ce qui n'est qu'une approximation puisque ces valeurs sont déterminées expérimentalement. Toutefois, l'expérience montre que cette approximation est valable.
Les diverses options de cette fenêtre sont accessibles directement au clavier, à l'aide des raccourcis suivants :
| Touche | Commande |
|---|---|
| « espace » | fermer la fenêtre |
| 1 | valeur de E1 |
| Shift + 1 | valeur de E'1 (en mode « Deux points ») |
| - | diminuer le degré du polynôme |
| + | augmenter le degré du polynôme |
| d | utiliser un second point de contrôle (E'1) |
Pour comparer divers spectres d'absorption X, il est nécessaire de les normaliser. La convention usuelle est de supposer que la hauteur de seuil pour l'atome isolé vaut 1, soit µ0(E0) = 1. Cette option permet de réaliser cette normalisation. À l'appel de l'option, le programme ouvre une fenêtre qui permet de choisir la façon dont est modélisée la hauteur de seuil.

Comme le but est principalement la comparaison des spectres, il est possible d'afficher simultanément un spectre déjà normalisé (il apparaît alors en gris foncé), à l'échelle du spectre en cours de normalisation après normalisation dans les conditions choisies, de façon à optimiser cette normalisation. Pour cela, cliquez sur le bouton à côté de Ref : et choisissez le spectre de référence à afficher (un clic sur Annuler, au lieu de choisir un graphe, conduit à ne plus afficher de référence).
LASE dispose de trois méthodes pour réaliser cette normalisation : en utilisant un ou deux points, ou par un modèle de µ0.
La normalisation est effectuée de telle sorte que le point indiqué (par la barre verticale rouge) vaille 1 après normalisation ; l'ancien minimum du spectre vaut 0.
La normalisation est effectuée de telle sorte que l'un des points indiqués vaille 1, l'autre 0 après normalisation. Le point qui vaudra 0 est repéré en vert, le point qui vaudra 1 est repéré en rouge.
Le programme modélise le coefficient d'absorption de l'atome absorbeur supposé isolé, µ0. Il faut lui indiquer à quelle énergie se situe le seuil : par définition, à cette énergie, µ0 = 1. Le programme normalise alors le signal expérimental pour que le signal vaille 1 à cette énergie et 0 au minimum. Cette méthode de normalisation est la plus rigoureuse en général.
Le coefficient d'absorption est modélisé comme pour la première étape de l'extraction des oscillations E.X.A.F.S. Le trait vert représente la position de E2, il peut être déplacé grâce à un clic du bouton droit de la souris ; le trait rouge indique la position de l'énergie servant pour la normalisation (énergie E pour laquelle, par hypothèse, µ0(E) = 1), il peut être déplacé avec le bouton gauche de la souris. Le modèle de µ0 est tracé en bleu.
Lorsque les conditions de normalisation vous satisfont, il faut fermer la fenêtre pour les valider. Apparaît alors un message qui vous demande que faire (ci-dessous).

Annuler annule la fermeture, Fermer ferme la fenêtre sans faire le calcul, Ce spectre normalise le spectre traité uniquement et Série normalise tous les spectres de même type appartenant à la même série. Dans ce cas, les paramètres de la normalisation sont optimisés pour chaque spectre de sorte que, après normalisation, les différents spectres soient aussi superposables que possible (distance minimale au sens des moindres carrés).
Si le bouton Nouveau graphe est actif, le spectre original est conservé, sa version normalisée est calculée dans un nouveau graphe, de même nom avec la mention « normalisé ». Si le graphe original contient des barres d'erreur, elles sont adaptées lors de la normalisation (multiplication par le facteur d'échelle, ce qui suppose qu'il est parfaitement connu).
Cette option calcule la dérivée du coefficient d'absorption en utilisant la méthode des splines : le calcul de la spline qui modélise le graphe proposé donne une famille de coefficients (ai, bi, ci) tels que, si (xi, yi) est un point du spectre :
si xi < x < xi + 1, alors y = yi + ai(x - xi) + bi(x - xi)2 + ci(x - xi)3.
Dans ce cas, la dérivée est estimée par :
y' = ai + 2bi(x - xi) + 3ci (x - xi)2.
Cette option calcule la dérivée seconde du coefficient d'absorption, en utilisant la méthode des splines. Avec les notations précédentes, la dérivée seconde est choisie égale à : y'' = 2bi + 6ci(x - xi)
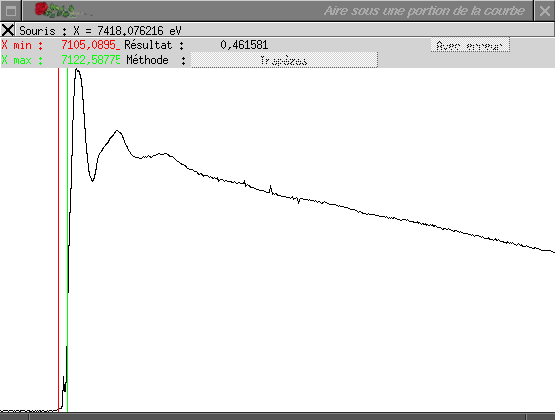
Cette option permet de calculer l'aire définie par une portion d'un graphe &emdash; elle permet donc, en particulier, de calculer la surface d'un pic du préseuil d'un spectre d'absorption des rayons X. La fenêtre qui s'ouvre permet de définir la région à intégrer : la barre rouge indique la borne inférieure (elle se place par un clic avec le premier bouton de la souris ou en cliquant sur sa valeur) ; la barre verte indique la borne supérieure (elle se place par un clic avec le deuxième bouton de la souris ou en cliquant sur sa valeur). Le résultat est affiché en temps réel dans la partie haute de la fenêtre.
Dans sa version actuelle, l'intégration est réalisée en utilisant la méthode des trapèzes : si les points du spectre compris entre xmin et xmax sont les points d'indice i à i + n, alors l'intégrale est estimée par

| Le menu « graphes » (mode extraction) | Sommaire | Le menu « EXAFS » (mode extraction) |